



浩浩(化名)是个非常可爱的孩子,可是近期妈妈发现浩浩的个子不长了,肚子越来越大,长相也有些和以前不同,手脚越发不灵活。于是妈妈带着浩浩经过多方检查,最终基因检测明确了浩浩是黏多糖贮积症II型患者。
罕见病--黏多糖贮积症
黏多糖贮积症II型(MPS),又称为亨特综合征,是一种罕见的遗传性代谢疾病,由于体内缺乏艾杜糖醛酸-2-硫酸酯酶,导致黏多糖无法正常分解,进而引发一系列严重的生理问题。
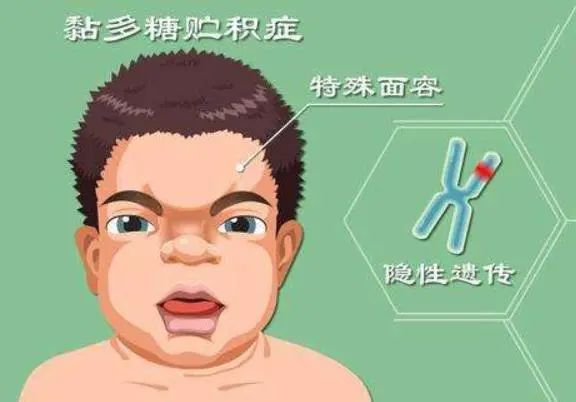
MPSII型患者出生时多表现正常,随着年龄增长症状逐渐明显,表现出典型的特殊面容、骨关节异常、身材矮小、皮肤改变、肝脾肿大以及神经系统、呼吸系统、心血管系统、眼部、耳鼻喉等多器官系统受累的症状,严重者常在青少年时期因神经系统退化和心肺功能衰竭而死亡,难以存活到成年。
本病虽然罕见,但并非不治之症,特异性治疗包括酶替代治疗和造血干细胞移植治疗,其中酶替代治疗药物已通过国家药监局快速审批通道获批,于2020年9月在我国上市。
首例替代治疗
浩浩妈妈通过多方打听,慕名来到江西省儿童医院内分泌遗传代谢科求助。入科后,内分泌遗传代谢科医疗团队对浩浩的病情反复评估,对知情同意、用药审批、输注流程、应急预案等做了充分准备。

科室团队严格按照《黏多糖贮积症Ⅱ型临床诊断与治疗专家共识》进行详细的临床检查与治疗前评估,制定缜密的用药计划。经过医护团队充分准备,药物成功输入,整个输注过程历时2个多小时,未出现不良药物反应,全程生命体征平稳,初步显示出治疗的良好效果。
该案例的成功实践,不仅填补了医院在黏多糖贮积症II型酶替代治疗领域的空白,也进一步验证了酶替代疗法在罕见病治疗中的重要作用。未来,医院将继续加大对罕见病诊疗技术的研发与应用力度,致力于提供更全面、更精准、更高效的医疗服务,让每一位罕见病患者都能得到及时有效的治疗,共享健康生活。
请输入验证码